Ligand

Cobalt complex HCo(CO)4 with five ligands
| Look up ligand in Wiktionary, the free dictionary. |
In coordination chemistry, a ligand[help 1] is an ion or molecule (functional group) that binds to a central metal atom to form a coordination complex. The bonding with the metal generally involves formal donation of one or more of the ligand's electron pairs. The nature of metal–ligand bonding can range from covalent to ionic. Furthermore, the metal–ligand bond order can range from one to three. Ligands are viewed as Lewis bases, although rare cases are known to involve Lewis acidic "ligands".[1][2]
Metals and metalloids are bound to ligands in virtually all circumstances, although gaseous "naked" metal ions can be generated in high vacuum. Ligands in a complex dictate the reactivity of the central atom, including ligand substitution rates, the reactivity of the ligands themselves, and redox. Ligand selection is a critical consideration in many practical areas, including bioinorganic and medicinal chemistry, homogeneous catalysis, and environmental chemistry.
Ligands are classified in many ways, including: charge, size (bulk), the identity of the coordinating atom(s), and the number of electrons donated to the metal (denticity or hapticity). The size of a ligand is indicated by its cone angle.
Contents
1 History
2 Strong field and weak field ligands
3 Classification of ligands as L and X
4 Polydentate and polyhapto ligand motifs and nomenclature
4.1 Denticity
4.2 Hapticity
5 Ligand motifs
5.1 Trans-spanning ligands
5.2 Ambidentate ligand
5.3 Bridging ligand
5.4 Binucleating ligand
5.5 Metal–ligand multiple bond
5.6 Spectator ligand
5.7 Bulky ligands
5.8 Chiral ligands
5.9 Hemilabile ligands
5.10 Non-innocent ligand
6 Common ligands
6.1 Examples of common ligands (by field strength)
6.2 Other generally encountered ligands (alphabetical)
7 Ligand exchange
8 Ligand–protein binding database
9 See also
10 Notes
11 References
12 External links
History
The composition of coordination complexes have been known since the early 1800s, such as Prussian blue and copper vitriol. The key breakthrough occurred when Alfred Werner reconciled formulas and isomers. He showed, among other things, that the formulas of many cobalt(III) and chromium(III) compounds can be understood if the metal has six ligands in an octahedral geometry. The first to use the term "ligand" were Alfred Stock and Carl Somiesky, in relation to silicon chemistry. The theory allows one to understand the difference between coordinated and ionic chloride in the cobalt ammine chlorides and to explain many of the previously inexplicable isomers. He resolved the first coordination complex called hexol into optical isomers, overthrowing the theory that chirality was necessarily associated with carbon compounds.[3][4]
Strong field and weak field ligands
In general, ligands are viewed as electron donors and the metals as electron acceptors. This is because the ligand and central metal are bonded to one another, and the ligand is providing both electrons to the bond (lone pair of electrons) instead of the metal and ligand each providing one electron. Bonding is often described using the formalisms of molecular orbital theory. The HOMO (Highest Occupied Molecular Orbital) can be mainly of ligands or metal character.
Ligands and metal ions can be ordered in many ways; one ranking system focuses on ligand 'hardness' (see also hard/soft acid/base theory). Metal ions preferentially bind certain ligands. In general, 'hard' metal ions prefer weak field ligands, whereas 'soft' metal ions prefer strong field ligands. According to the molecular orbital theory, the HOMO of the ligand should have an energy that overlaps with the LUMO (Lowest Unoccupied Molecular Orbital) of the metal preferential. Metal ions bound to strong-field ligands follow the Aufbau principle, whereas complexes bound to weak-field ligands follow Hund's rule.
Binding of the metal with the ligands results in a set of molecular orbitals, where the metal can be identified with a new HOMO and LUMO (the orbitals defining the properties and reactivity of the resulting complex) and a certain ordering of the 5 d-orbitals (which may be filled, or partially filled with electrons). In an octahedral environment, the 5 otherwise degenerate d-orbitals split in sets of 2 and 3 orbitals (for a more in depth explanation, see crystal field theory).
- 3 orbitals of low energy: dxy, dxz and dyz
- 2 of high energy: dz2 and dx2−y2
- 3 orbitals of low energy: dxy, dxz and dyz
The energy difference between these 2 sets of d-orbitals is called the splitting parameter, Δo. The magnitude of Δo is determined by the field-strength of the ligand: strong field ligands, by definition, increase Δo more than weak field ligands. Ligands can now be sorted according to the magnitude of Δo (see the table below). This ordering of ligands is almost invariable for all metal ions and is called spectrochemical series.
For complexes with a tetrahedral surrounding, the d-orbitals again split into two sets, but this time in reverse order.
- 2 orbitals of low energy: dz2 and dx2−y2
- 3 orbitals of high energy: dxy, dxz and dyz
- 2 orbitals of low energy: dz2 and dx2−y2
The energy difference between these 2 sets of d-orbitals is now called Δt. The magnitude of Δt is smaller than for Δo, because in a tetrahedral complex only 4 ligands influence the d-orbitals, whereas in an octahedral complex the d-orbitals are influenced by 6 ligands. When the coordination number is neither octahedral nor tetrahedral, the splitting becomes correspondingly more complex. For the purposes of ranking ligands, however, the properties of the octahedral complexes and the resulting Δo has been of primary interest.
The arrangement of the d-orbitals on the central atom (as determined by the 'strength' of the ligand), has a strong effect on virtually all the properties of the resulting complexes. E.g., the energy differences in the d-orbitals has a strong effect in the optical absorption spectra of metal complexes. It turns out that valence electrons occupying orbitals with significant 3 d-orbital character absorb in the 400–800 nm region of the spectrum (UV–visible range). The absorption of light (what we perceive as the color) by these electrons (that is, excitation of electrons from one orbital to another orbital under influence of light) can be correlated to the ground state of the metal complex, which reflects the bonding properties of the ligands. The relative change in (relative) energy of the d-orbitals as a function of the field-strength of the ligands is described in Tanabe–Sugano diagrams.
In cases where the ligand has low energy LUMO, such orbitals also participate in the bonding. The metal–ligand bond can be further stabilised by a formal donation of electron density back to the ligand in a process known as back-bonding. In this case a filled, central-atom-based orbital donates density into the LUMO of the (coordinated) ligand. Carbon monoxide is the preeminent example a ligand that engages metals via back-donation. Complementarily, ligands with low-energy filled orbitals of pi-symmetry can serve as pi-donor.

Metal–EDTA complex, wherein the aminocarboxylate is a hexadentate (chelating) ligand.

Cobalt(III) complex containing six ammonia ligands, which are monodentate. The chloride is not a ligand.
Classification of ligands as L and X
Especially in the area of organometallic chemistry, ligands are classified as L and X (or combinations of the two). The classification scheme – the "CBC Method" for Covalent Bond Classification – was popularized by M.L.H. Green and "is based on the notion that there are three basic types [of ligands]... represented by the symbols L, X, and Z, which correspond respectively to 2-electron, 1-electron and 0-electron neutral ligands."[5][6] Another type of ligand worthy of consideration is the LX ligand which as expected from the used conventional representation will donate three electrons if NVE (Number of Valence Electrons) required. Example is alkoxy ligands( which is regularly known as X ligand too). L ligands are derived from charge-neutral precursors and are represented by amines, phosphines, CO, N2, and alkenes. X ligands typically are derived from anionic precursors such as chloride but includes ligands where salts of anion do not really exist such as hydride and alkyl. Thus, the complex IrCl(CO)(PPh3)2 is classified as an MXL3 complex, since CO and the two PPh3 ligands are classified as Ls. The oxidative addition of H2 to IrCl(CO)(PPh3)2 gives an 18e− ML3X3 product, IrClH2(CO)(PPh3)2. EDTA4− is classified as an L2X4 ligand, as it features four anions and two neutral donor sites. Cp is classified as an L2X ligand.[7]
Polydentate and polyhapto ligand motifs and nomenclature
Denticity
Denticity (represented by κ) refers to the number of times a ligand bonds to a metal through noncontiguous donor sites. Many ligands are capable of binding metal ions through multiple sites, usually because the ligands have lone pairs on more than one atom. Ligands that bind via more than one atom are often termed chelating. A ligand that binds through two sites is classified as bidentate, and three sites as tridentate. The "bite angle" refers to the angle between the two bonds of a bidentate chelate. Chelating ligands are commonly formed by linking donor groups via organic linkers. A classic bidentate ligand is ethylenediamine, which is derived by the linking of two ammonia groups with an ethylene (−CH2CH2−) linker. A classic example of a polydentate ligand is the hexadentate chelating agent EDTA, which is able to bond through six sites, completely surrounding some metals. The number of times a polydentate ligand binds to a metal centre is symbolized by "κn", where n indicates the number of sites by which a ligand attaches to a metal. EDTA4−, when it is hexidentate, binds as a κ6-ligand, the amines and the carboxylate oxygen atoms are not contiguous. In practice, the n value of a ligand is not indicated explicitly but rather assumed. The binding affinity of a chelating system depends on the chelating angle or bite angle.
Complexes of polydentate ligands are called chelate complexes. They tend to be more stable than complexes derived from monodentate ligands. This enhanced stability, the chelate effect, is usually attributed to effects of entropy, which favors the displacement of many ligands by one polydentate ligand. When the chelating ligand forms a large ring that at least partially surrounds the central atom and bonds to it, leaving the central atom at the centre of a large ring. The more rigid and the higher its denticity, the more inert will be the macrocyclic complex. Heme is a good example: the iron atom is at the centre of a porphyrin macrocycle, being bound to four nitrogen atoms of the tetrapyrrole macrocycle. The very stable dimethylglyoximate complex of nickel is a synthetic macrocycle derived from the anion of dimethylglyoxime.
Hapticity
Hapticity (represented by η) refers to the number of contiguous atoms that comprise a donor site and attach to a metal center. Butadiene forms both η2 and η4 complexes depending on the number of carbon atoms that are bonded to the metal.[7]
Ligand motifs
Trans-spanning ligands
Trans-spanning ligands are bidentate ligands that can span coordination positions on opposite sides of a coordination complex.[8]
Ambidentate ligand
Unlike polydentate ligands, ambidentate ligands can attach to the central atom in two places. A good example of this is thiocyanate, SCN−, which can attach at either the sulfur atom or the nitrogen atom. Such compounds give rise to linkage isomerism. Polyfunctional ligands, see especially proteins, can bond to a metal center through different ligand atoms to form various isomers.
Bridging ligand
A bridging ligand links two or more metal centers. Virtually all inorganic solids with simple formulas are coordination polymers, consisting of metal ion centres linked by bridging ligands. This group of materials includes all anhydrous binary metal ion halides and pseudohalides. Bridging ligands also persist in solution. Polyatomic ligands such as carbonate are ambidentate and thus are found to often bind to two or three metals simultaneously. Atoms that bridge metals are sometimes indicated with the prefix "μ". Most inorganic solids are polymers by virtue of the presence of multiple bridging ligands. Bridging ligands, capable of coordinating multiple metal ions, have been attracting considerable interest because of their potential use as building blocks for the fabrication of functional multimetallic assemblies.[9]
Binucleating ligand
Binucleating ligands bind two metals.[10] Usually binucleating ligands feature bridging ligands, such as phenoxide, pyrazolate, or pyrazine, as well as other donor groups that bind to only one of the two metals.
Metal–ligand multiple bond
Some ligands can bond to a metal center through the same atom but with a different number of lone pairs. The bond order of the metal ligand bond can be in part distinguished through the metal ligand bond angle (M−X−R). This bond angle is often referred to as being linear or bent with further discussion concerning the degree to which the angle is bent. For example, an imido ligand in the ionic form has three lone pairs. One lone pair is used as a sigma X donor, the other two lone pairs are available as L-type pi donors. If both lone pairs are used in pi bonds then the M−N−R geometry is linear. However, if one or both these lone pairs is nonbonding then the M−N−R bond is bent and the extent of the bend speaks to how much pi bonding there may be. η1-Nitric oxide can coordinate to a metal center in linear or bent manner.
Spectator ligand
A spectator ligand is a tightly coordinating polydentate ligand that does not participate in chemical reactions but removes active sites on a metal. Spectator ligands influence the reactivity of the metal center to which they are bound.
Bulky ligands
Bulky ligands are used to control the steric properties of a metal center. They are used for many reasons, both practical and academic. On the practical side, they influence the selectivity of metal catalysts, e.g., in hydroformylation. Of academic interest, bulky ligands stabilize unusual coordination sites, e.g., reactive coligands or low coordination numbers. Often bulky ligands are employed to simulate the steric protection afforded by proteins to metal-containing active sites. Of course excessive steric bulk can prevent the coordination of certain ligands.
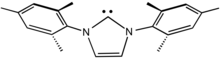
The N-heterocyclic carbene ligand called IMes is a bulky ligand by virtue of the pair of mesityl groups.
Chiral ligands
Chiral ligands are useful for inducing asymmetry within the coordination sphere. Often the ligand is employed as an optically pure group. In some cases, such as secondary amines, the asymmetry arises upon coordination. Chiral ligands are used in homogeneous catalysis, such as asymmetric hydrogenation.
Hemilabile ligands
Hemilabile ligands contain at least two electronically different coordinating groups and form complexes where one of these is easily displaced from the metal center while the other remains firmly bound, a behaviour which has been found to increase the reactivity of catalysts when compared to the use of more traditional ligands.
Non-innocent ligand
Non-innocent ligands bond with metals in such a manner that the distribution of electron density between the metal center and ligand is unclear. Describing the bonding of non-innocent ligands often involves writing multiple resonance forms that have partial contributions to the overall state.
Common ligands
Virtually every molecule and every ion can serve as a ligand for (or "coordinate to") metals. Monodentate ligands include virtually all anions and all simple Lewis bases. Thus, the halides and pseudohalides are important anionic ligands whereas ammonia, carbon monoxide, and water are particularly common charge-neutral ligands. Simple organic species are also very common, be they anionic (RO− and RCO−
2) or neutral (R2O, R2S, R3−xNHx, and R3P). The steric properties of some ligands are evaluated in terms of their cone angles.
Beyond the classical Lewis bases and anions, all unsaturated molecules are also ligands, utilizing their pi electrons in forming the coordinate bond. Also, metals can bind to the σ bonds in for example silanes, hydrocarbons, and dihydrogen (see also: Agostic interaction).
In complexes of non-innocent ligands, the ligand is bonded to metals via conventional bonds, but the ligand is also redox-active.
Examples of common ligands (by field strength)
In the following table the ligands are sorted by field strength (weak field ligands first):
| Ligand | formula (bonding atom(s) in bold) | Charge | Most common denticity | Remark(s) |
|---|---|---|---|---|
Iodide (iodo) | I− | monoanionic | monodentate | |
Bromide (bromido) | Br− | monoanionic | monodentate | |
Sulfide (thio or less commonly "bridging thiolate") | S2− | dianionic | monodentate (M=S), or bidentate bridging (M−S−M′) | |
Thiocyanate (S-thiocyanato) | S−CN− | monoanionic | monodentate | ambidentate (see also isothiocyanate, below) |
Chloride (chlorido) | Cl− | monoanionic | monodentate | also found bridging |
Nitrate (nitrato) | O−NO− 2 | monoanionic | monodentate | |
Azide (azido) | N−N− 2 | monoanionic | monodentate | Very Toxic |
Fluoride (fluoro) | F− | monoanionic | monodentate | |
Hydroxide (hydroxido) | O−H− | monoanionic | monodentate | often found as a bridging ligand |
Oxalate (oxalato) | [O−CO−CO−O]2− | dianionic | bidentate | |
Water (aqua) | O−H2 | neutral | monodentate | |
Nitrite (nitrito) | O−N−O− | monoanionic | monodentate | ambidentate (see also nitro) |
Isothiocyanate (isothiocyanato) | N=C=S− | monoanionic | monodentate | ambidentate (see also thiocyanate, above) |
Acetonitrile (acetonitrilo) | CH3CN | neutral | monodentate | |
Pyridine (py) | C5H5N | neutral | monodentate | |
Ammonia (ammine or less commonly "ammino") | NH3 | neutral | monodentate | |
Ethylenediamine (en) | NH2−CH2−CH2−NH2 | neutral | bidentate | |
2,2′-Bipyridine (bipy) | NC5H4−C5H4N | neutral | bidentate | easily reduced to its (radical) anion or even to its dianion |
1,10-Phenanthroline (phen) | C12H8N2 | neutral | bidentate | |
Nitrite (nitro) | N−O− 2 | monoanionic | monodentate | ambidentate (see also nitrito) |
| Triphenylphosphine | P−(C6H5)3 | neutral | monodentate | |
Cyanide (cyano) | C≡N− N≡C− | monoanionic | monodentate | can bridge between metals (both metals bound to C, or one to C and one to N) |
Carbon monoxide (carbonyl) | –CO, others | neutral | monodentate | can bridge between metals (both metals bound to C) |
The entries in the table are sorted by field strength, binding through the stated atom (i.e. as a terminal ligand). The 'strength' of the ligand changes when the ligand binds in an alternative binding mode (e.g., when it bridges between metals) or when the conformation of the ligand gets distorted (e.g., a linear ligand that is forced through steric interactions to bind in a nonlinear fashion).
Other generally encountered ligands (alphabetical)
In this table other common ligands are listed in alphabetical order.
| Ligand | formula (bonding atom(s) in bold) | Charge | Most common denticity | Remark(s) |
|---|---|---|---|---|
Acetylacetonate (acac) | CH3−CO−CH2−CO−CH3 | monoanionic | bidentate | In general bidentate, bound through both oxygens, but sometimes bound through the central carbon only, see also analogous ketimine analogues |
| Alkenes | R2C=CR2 | neutral | compounds with a C−C double bond | |
Aminopolycarboxylic acids (APCAs) | | |||
BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid) | | |||
| Benzene | C6H6 | neutral | and other arenes | |
1,2-Bis(diphenylphosphino)ethane (dppe) | (C6H5)2P−C2H4−P(C6H5)2 | neutral | bidentate | |
1,1-Bis(diphenylphosphino)methane (dppm) | (C6H5)2P−CH2−P(C6H5)2 | neutral | Can bond to two metal atoms at once, forming dimers | |
| Corroles | tetradentate | |||
| Crown ethers | neutral | primarily for alkali and alkaline earth metal cations | ||
| 2,2,2-cryptand | hexadentate | primarily for alkali and alkaline earth metal cations | ||
| Cryptates | neutral | |||
Cyclopentadienyl (Cp) | C 5H− 5 | monoanionic | Although monoanionic, by the nature of its occupied molecular orbitals, it is capable of acting as a tridentate ligand. | |
Diethylenetriamine (dien) | C4H13N3 | neutral | tridentate | related to TACN, but not constrained to facial complexation |
Dimethylglyoximate (dmgH−) | monoanionic | |||
1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) | | |||
| Diethylenetriaminepentaacetic acid (DTPA) (pentetic acid) | | |||
Ethylenediaminetetraacetic acid (EDTA) (edta4−) | (−OOC−CH2)2N−C2H4−N(CH2-COO−)2 | tetraanionic | hexadentate | |
| Ethylenediaminetriacetate | −OOC−CH2NH−C2H4−N(CH2-COO−)2 | trianionic | pentadentate | |
Ethyleneglycolbis(oxyethylenenitrilo)tetraacetate (egta4−) | (−OOC−CH2)2N−C2H4−O−C2H4−O−C2H4−N(CH2−COO−)2 | tetraanionic | octodentate | |
| Fura-2 | | |||
Glycinate (glycinato) | NH2CH2COO− | monoanionic | bidentate | other α-amino acid anions are comparable (but chiral) |
| Heme | dianionic | tetradentate | macrocyclic ligand | |
Iminodiacetic acid (IDA) | tridentate | Used extensively to make radiotracers for scintigraphy by complexing the metastable radionuclide technetium-99m. For example, in cholescintigraphy, HIDA, BrIDA, PIPIDA, and DISIDA are used | ||
| Nicotianamine | Ubiquitous in higher plants | |||
| Nitrosyl | NO+ | cationic | bent (1e−) and linear (3e−) bonding mode | |
Nitrilotriacetic acid (NTA) | | |||
| Oxo | O2− | dianion | monodentate | sometimes bridging |
| Pyrazine | N2C4H4 | neutral | ditopic | sometimes bridging |
| Scorpionate ligand | tridentate | |||
| Sulfite | O−SO2− 2 S−O2− 3 | monoanionic | monodentate | ambidentate |
2,2′;6′,2″-Terpyridine (terpy) | NC5H4−C5H3N−C5H4N | neutral | tridentate | meridional bonding only |
Triazacyclononane (tacn) | (C2H4)3(NR)3 | neutral | tridentate | macrocyclic ligand see also the N,N′,N″-trimethylated analogue |
| Tricyclohexylphosphine | P(C6H11)3 or PCy3 | neutral | monodentate | |
Triethylenetetramine (trien) | C6H18N4 | neutral | tetradentate | |
| Trimethylphosphine | P(CH3)3 | neutral | monodentate | |
| Tri(o-tolyl)phosphine | P(o-tolyl)3 | neutral | monodentate | |
Tris(2-aminoethyl)amine (tren) | (NH2CH2CH2)3N | neutral | tetradentate | |
| Tris(2-diphenylphosphineethyl)amine (np3) | neutral | tetradentate | ||
| Tropylium | C 7H+ 7 | cationic | ||
| Carbon dioxide | –CO2, others | neutral | see metal carbon dioxide complex | |
Phosphorus trifluoride (trifluorophosphorus) | –PF3 | neutral |
Ligand exchange
A ligand exchange (also ligand substitution) is a type of chemical reaction in which a ligand in a compound is replaced by another. One type of pathway for substitution is the ligand dependent pathway. In organometallic chemistry this can take place via associative substitution or by dissociative substitution. Another form of ligand exchange is seen in the nucleophilic abstraction reaction.
Ligand–protein binding database
BioLiP is a comprehensive ligand–protein interaction database, with the 3D structure of the ligand–protein interactions taken from the Protein Data Bank. MANORAA is a webserver for analyzing conserved and differential molecular interaction of the ligand in complex with protein structure homologs from the Protein Data Bank. It provides the linkage to protein targets such as its location in the biochemical pathways, SNPs and protein/RNA baseline expression in target organ.[11]
See also
- Bridging carbonyl
- Coordination complex
- Crystal field theory
- DNA binding ligand
- Inorganic chemistry
- Josiphos ligands
- Ligand dependent pathway
- Ligand field theory
- Ligand isomerism
- Spectrochemical series
- Tanabe–Sugano diagram
Notes
^ The word ligand comes from Latin ligare, to bind/tie. It is pronounced either /ˈlaɪɡənd/ or /ˈlɪɡənd/; both are very common.
References
^ Cotton, Frank Albert; Geoffrey Wilkinson; Carlos A. Murillo (1999). Advanced Inorganic Chemistry. Wiley-Interscience. p. 1355. ISBN 978-0471199571..mw-parser-output cite.citation{font-style:inherit}.mw-parser-output q{quotes:"""""""'""'"}.mw-parser-output code.cs1-code{color:inherit;background:inherit;border:inherit;padding:inherit}.mw-parser-output .cs1-lock-free a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-limited a,.mw-parser-output .cs1-lock-registration a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-subscription a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration{color:#555}.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration span{border-bottom:1px dotted;cursor:help}.mw-parser-output .cs1-hidden-error{display:none;font-size:100%}.mw-parser-output .cs1-visible-error{font-size:100%}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-format{font-size:95%}.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-left{padding-left:0.2em}.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-right{padding-right:0.2em}
^ Miessler, Gary L.; Paul J. Fischer; Donald Arthur Tarr (2013). Inorganic Chemistry. Prentice Hall. p. 696. ISBN 978-0321811059.
^ Jackson, W. Gregory; Josephine A. McKeon; Silvia Cortez (1 October 2004). "Alfred Werner's Inorganic Counterparts of Racemic and Mesomeric Tartaric Acid: A Milestone Revisited". Inorganic Chemistry. 43 (20): 6249–6254. doi:10.1021/ic040042e. PMID 15446870.
^ Bowman-James, Kristin (2005). "Alfred Werner Revisited: The Coordination Chemistry of Anions". Accounts of Chemical Research. 38 (8): 671–678. doi:10.1021/ar040071t. PMID 16104690.
^ Green, M. L. H. (20 September 1995). "A new approach to the formal classification of covalent compounds of the elements". Journal of Organometallic Chemistry. 500 (1–2): 127–148. doi:10.1016/0022-328X(95)00508-N. ISSN 0022-328X.
^ "mlxz plots – Columbia University", Columbia University, New York.
^ ab Hartwig, J. F. Organotransition Metal Chemistry, from Bonding to Catalysis; University Science Books: New York, 2010.
ISBN 1-891389-53-X
^ von Zelewsky, A. "Stereochemistry of Coordination Compounds" John Wiley: Chichester, 1995.
ISBN 047195599X.
^ Sauvage,
J.-P.; Collin, J.-P.; Chambron, J.-C.; Guillerez, S.; Coudret, C.; Balzani, V.; Barigelletti, F.; De Cola, L.; Flamigni, L. Chem. ReV. 1994, 94, 993-1019
^ Gavrilova, A. L.; Bosnich, B., "Principles of Mononucleating and Binucleating Ligand Design", Chem. Rev. 2004, volume 104, 349-383. doi:10.1021/cr020604g
^ Tanramluk D, Naripiyakul L, Akavipat R, Gong S, Charoensawan V (2016). "MANORAA (Mapping Analogous Nuclei Onto Residue And Affinity) for identifying protein-ligand fragment interaction, pathways and SNPs". Nucleic Acids Research: W514–21. doi:10.1093/nar/gkw314. PMC 4987895. PMID 27131358.
External links
| Wikimedia Commons has media related to Ligands. |
- See the modeling of ligand–receptor–ligand binding in Vu-Quoc, L., Configuration integral (statistical mechanics), 2008. this wiki site is down; see this article in the web archive on 2012 April 28.


